Introduction
The blood fluke Schistosoma is a parasitic trematode that is responsible for more than 200,000 human deaths per year. The state of the art to detect Schistosoma viability viability involves microscopy and knowledge of parasite morphology. The lack of appropriate methods for quantifying Schistosoma viability blocks the development of new anthelmintics.
In this application note, we present a fluorescence intensity-based microtiter plate assay to reproducibly detect schistosomal viability. The principle of the assay is based on differential membrane permeabilities of the dyes, fluorescein diacetate (FDA), and propidium iodide (PI). Fluorescein diacetate is able to cross the membrane of living cells. Once inside the cell, esterases will cut the diacetate, and fluorescein is released resulting in a measurable fluorescence signal that is directly related to the number of living cells. In contrast, propidium iodide cannot enter a viable cell. This dye will only stain the DNA of dead cells when the membrane has been compromised. The simultaneous detection of both propidium iodide and fluorescein diacetate measures allowed us to develop a fluorescence-based, microplate bioassay to improve detection of schistosomal viability. Using this flexible bioassay, we demonstrate its versatility in detecting schistosomal viability in response to a known inhibitor of thioredoxin glutathione reductase (auranofin).
Materials & Methods
- Black-walled, clear, and flat-bottomed 96-well plates from FisherScientific
- Black-walled, clear, and flat-bottomed 384-well plates from Matrix
Schistosomula preparation and culturing
Schistosoma infected snails were exposed to light. The snails shed cercariae that were subsequently converted to schistosomula by mechanical transformation. After purification, microscopic examination was done to assess quantity and quality of purified schistosomula. After preparation, the schistosomula were cultured in flasks for 24 hours and aliquoted into either 96-well plates (1000 parasites per 200 μL) or 384-well plates (200 parasites per 40 μL).
Schistosomula viability determination in response to auranofin
Purified schistosomula were cultured in microplates in phenol-red free DMEM for 24 h at 37°C and 5 % CO2 in presence of varying concentrations (10- 0.625 μM) of auranofin. After this, all schistosomula were washed three times to remove test compound and culture media supplements. After washing the parasites, propidium iodide and fluorescein diacetate were simultaneously added to each well to obtain a final concentration of 2 μg/mL and 0.5 μg/mL respectively.
Measurement in the BMG LABTECH microplate reader
Samples were measured from the bottom in dual chromatic mode with 544 nm excitation/620 nm emission to detect PI and 485 nm excitation/520 nm emission to detect FDA.
All fluorescent values were obtained with the plate reader incubator set at 37°C to ensure efficient esterase conversion of fluorescein diacetate to fluorescein within live schistosomula. Inclusion of appropriate control samples (live and heat-killed dead schistosomula) compensates for any inter-plate variations.
Data Handling
Numbers of live and dead schistosomula in each well were calculated using the following equations:
- LIVE (FDA fluorescence) = (Fsample – Fneg.contr.) / (Fpos.contr. – Fneg.contr)
- DEAD (PI fluorescence) = (Fsample – Fmedia control) / (Fneg.contr. – Fmedia control)
Schistosomula viability calculation:

Results & Discussion
With the help of a fluorescent microscope, we could confirm that FDA is useful for the staining of living cells and that PI is useful for the staining of dead cells. We could also confirm that no co-localization of FDA- or PI-devided fluorescence was observed in the same cell of an individual schistosomula.
Optimal fluorophore/schistosomula incubation time
In pretests, it was investigated how long both dyes should be incubated with the parasites in order to get maximal reproducible viability data. The results are shown in Fig. 1 and 2.
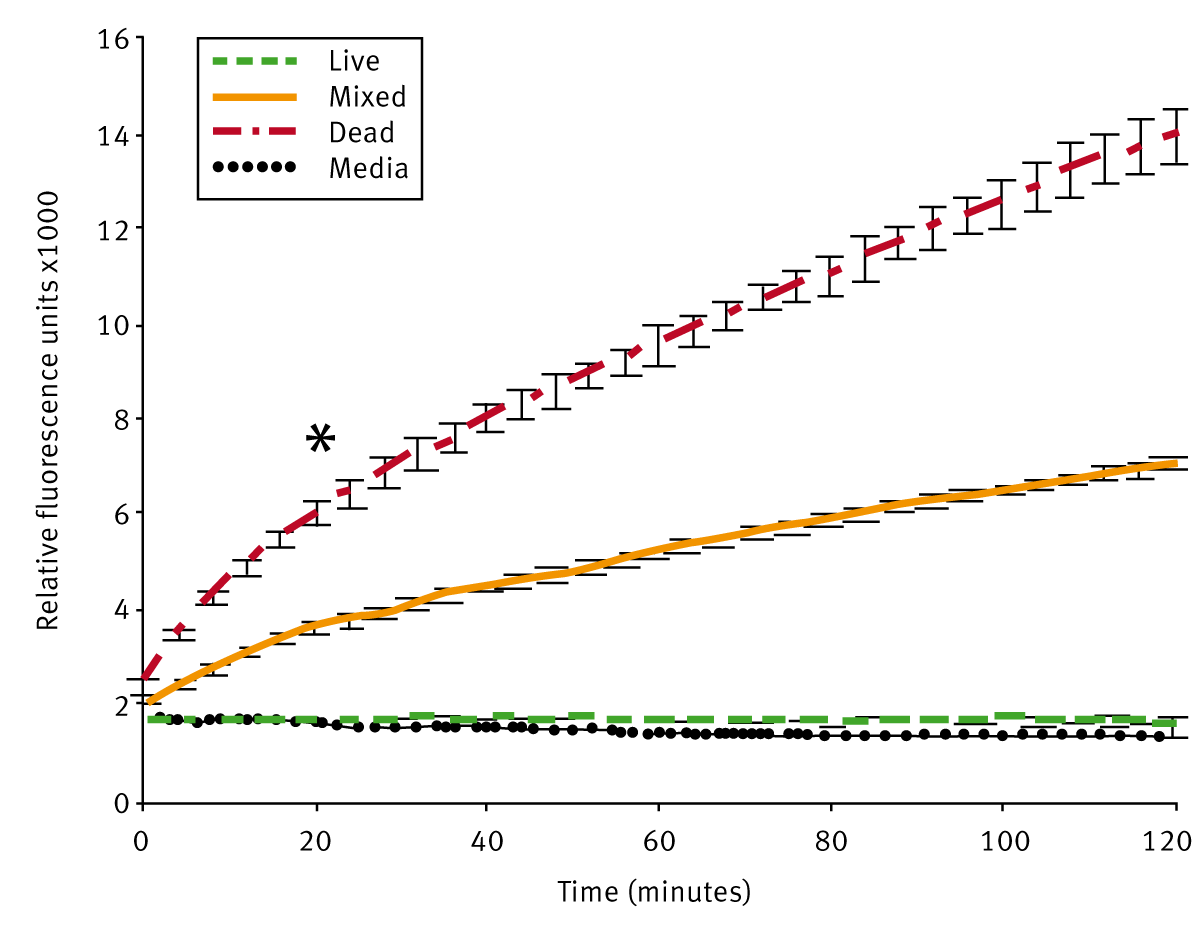
As expected dead parasites show the highest PI emission values whereas the signals for the live parasites are close to the media control and remain constant over time. The values for mixed samples containing live and dead schistosomula are in between. We chose an incubation time of 20 min to collect PI data as it provided an adequate time window to process multiple microtiter plates (* in Fig. 1). Whereas PI staining of dead schistosomula lead to an emission increase over at least 120 min, FDA staining showed different results (Fig. 2).
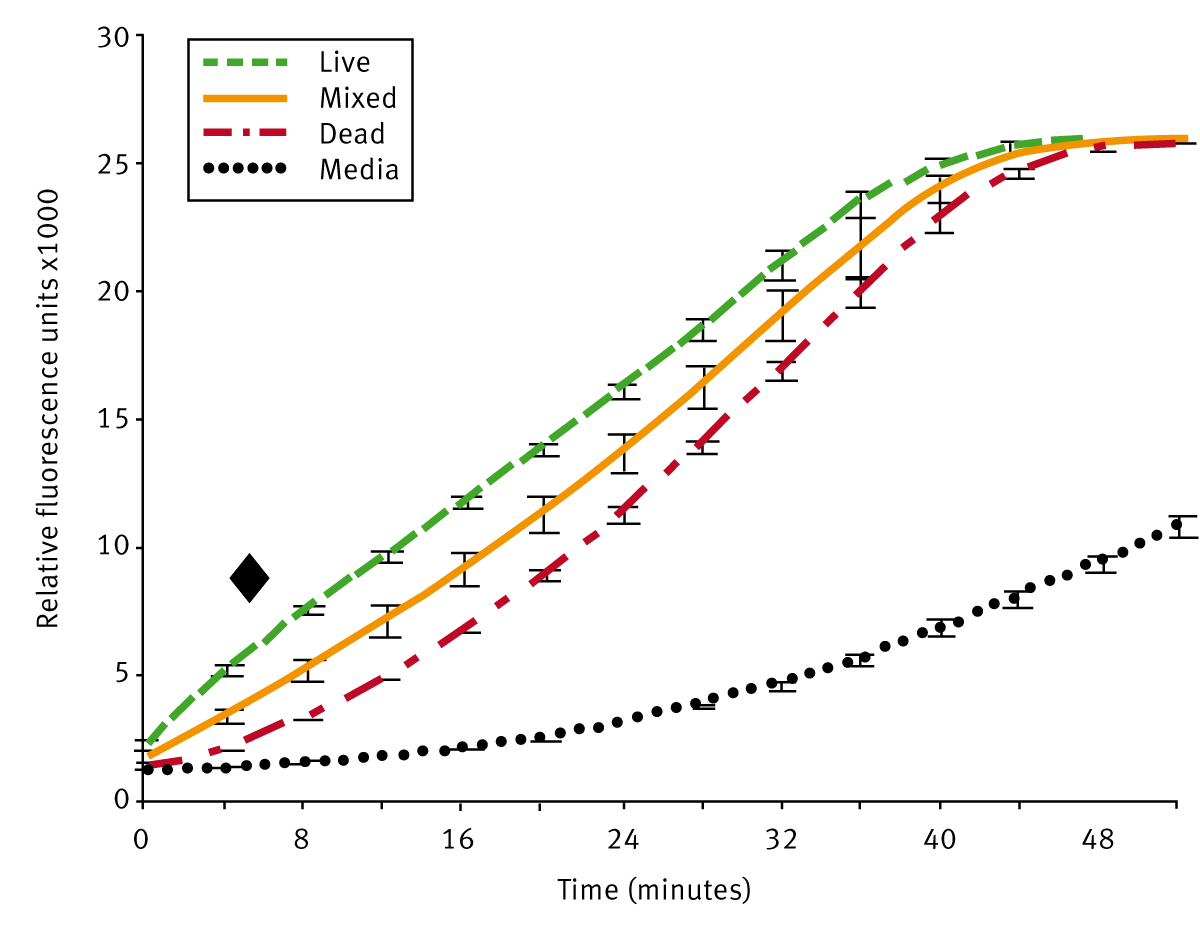
Live, dead, and mixed schistosomula populations generated emission data that quickly reached a plateau (51 min for 96-well plates). As expected the emission values for live schistosomula were highest, for dead lowest, and for the mixed population, we obtained intermediate values. The optimal FDA incubation time was calculated to be between 3 and 12 min. We chose 5 min to collect FDA data (Marker in Fig. 2).
Schistosomula viability in response to auranofin
To validate the assay, auranofin, an experimental TGR inhibitor, and anti-schistosomula compound, was added to the parasites during parasite growth. There is a clear and titratable anti-schistosomula effect on schistosomula viability (Fig. 3.
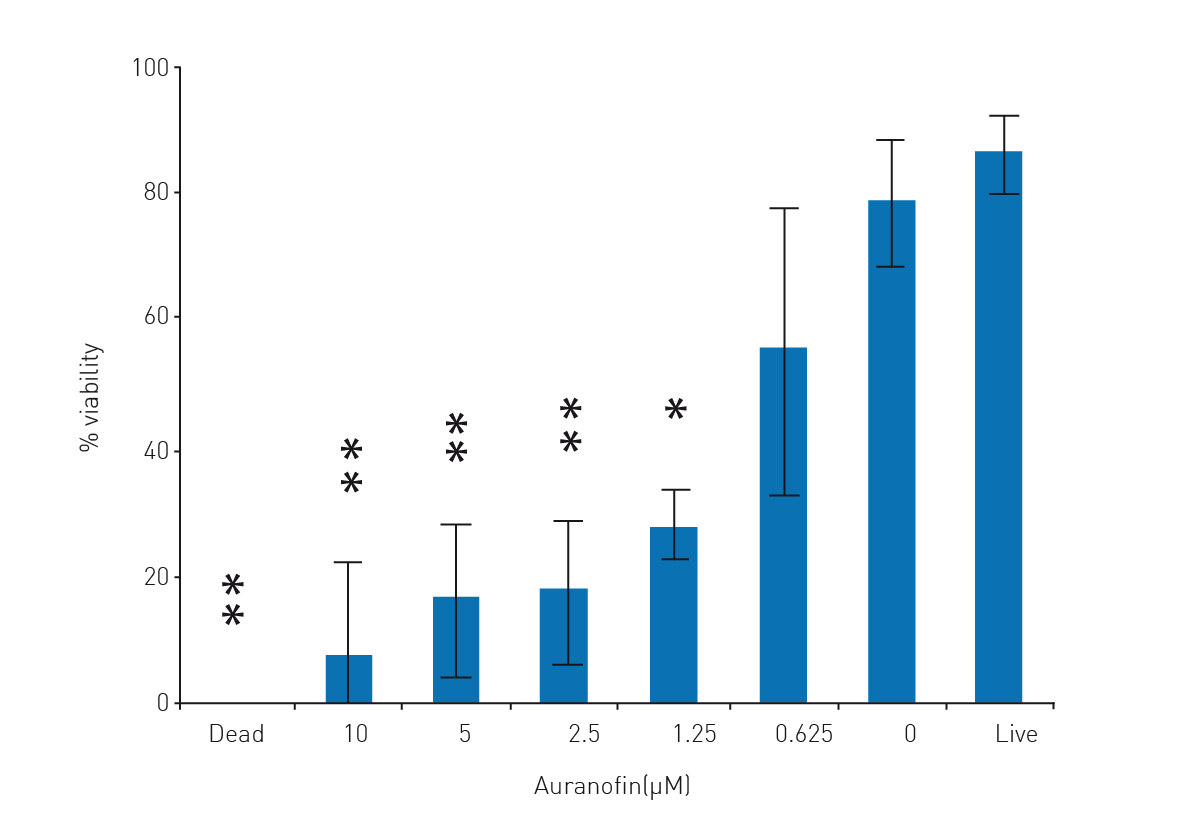
Percent viability transformations into probit values also allowed an auranofin LD50 calculation (0.82 ± 0.49 μM). Maximum drug effect was seen at 10 μM auranofin, where microscopic examination of schistosomula confirmed that death was 100 %.
Conclusion
In this application note, we demonstrate a quantitative, fast, and inexpensive method to reproducibly measure schistosomula viability. The use of a dual staining method is important for counteracting any pipetting errors and for verifying that differences in fluorescence are genuinely caused by differences in mortality.