Introduction
Ligand binding affinities at G-protein coupled receptors (GPCRs) have historically been determined using a radioligand that competes for receptor binding sites against an unlabelled drug-like compound. However, the potential hazards of opensource radioisotope handling, and the environmental impact of radioisotope disposal, make this a less desirable, costly technology. Development of fluorescent ligands for GPCRs provides a safer method for determining ligand binding affinities. However, quantification of fluorescent ligand binding tends to rely on high resolution fluorescence image capture. This approach can be lengthy and tedious, resulting in 96-well plate read times of approximately 30 minutes or more (depending on image number per well). Complex post-capture image analysis algorithms also extend data processing time. This results in High Content Analysis (HCA) assays that are too slow to be useful for High Throughput Screening (HTS).
In this study, we use the adjustable focal height feature and advanced bottom reading of the PHERAstar FS plate reader from BMG LABTECH, in a live cell 96-well plate-based assay, to quantify fluorescent ligand binding associated with the adherent cell layer at the bottom of the well. Real-time, multi-spot fluorescence intensity measurements and averaging per well, incorporated with blank subtraction and sample displacement curve calculations were performed ‘on the fly’ as the wells were being analysed. Depending on the well scan area, number of lamp flashes, and number of spots per well analysed, this resulted in all-inclusive 96-well plate ‘read to IC50 pKi curve plot’ times of between 2.5 and 10 minutes. This is in stark contrast to HCA assays, which can take around 60 minutes to achieve the same endpoint.
In this study, we illustrate the capability of the PHERAstar FS to perform fluorescent ligand binding assays using two of CellAura’s fluorescent ligands for adenosine and dopamine receptors. Furthermore, we postulate the suitability of such a combination of plate reader and fluorescent ligand for higher throughput receptor binding assays and screening.
Materials & Methods
The two fluorescent ligands used in these experiments were developed by CellAura Technologies Ltd. Ligand CA200645 is an adenosine A3 selective ligand, while CA200767 is a dopamine D1 selective ligand. Both ligands are labelled with the BODIPY 630/650 fluorophore that fluoresces at the far red end of the spectrum.
The A1-selective antagonist DPCPX and the A3-selective antagonist MRS1220 were obtained from Tocris. The D1-selective antagonist, SCH23390 was obtained from Sigma-Aldrich. Black-sided, clear-bottomed, 96-well view plates were obtained from Greiner Bio-One.
The PHERAstar FS was fitted with an optic module for BODIPY 630/650 to provide Excitation at 620 nm and measuring Emission at 660 nm.
Experimental
CHO cells expressing human adenosine A1 or A3 receptors, or dopamine D1 receptors, were grown to confluence in T75 flasks, harvested, and seeded into 96-well black view plates and incubated at 37°C, 5% CO2 for 24 hours prior to assay. On the day of assay, culture medium was aspirated and replaced with 100 μL serum-free media with or without an appropriate unlabelled competitor at 1.2x final concentration. Cells were incubated for 30 minutes at 37°C, 5% CO2. Fluorescent ligand at 6x final concentration was added in a 20 μL volume (at a 1:6 dilution) to the appropriate wells. Cells were incubated for a further 30 minutes at 37°C, 5% CO2. Cells were washed twice in HBS to remove unbound ligand, with a final 100 μL volume of HBS added to all wells.
Plates were read on the PHERAstar FS in bottom read mode using the 620/660 nm optic module. A 5x5 spot read matrix was selected to read from a 4 mm diameter in each well (a reduced diameter eliminates the edge of the well and any anomalies in the distribution of the cells in this region). Prior to reading the plate, a Z-height scan was performed to identify the optimal height for quantifying fluorescence bound to the cell layer at the bottom of the well. An automated gain adjustment was also performed, to give an optimal gain of 3300. With the above settings applied to the measurement a read time of 2.5 minutes per plate was possible.
Results & Discussion
The BODIPY 630/650-labelled fluorescent ligands bound to their target receptors and were detected using the PHERAstar FS. Mean fluorescence intensity differences between positive controls (fluorescent ligand without unlabelled competitor) and negative controls (without fluorescent ligand) were used to calculate assay robustness Z’ Factors. Binding of the fluorescent ligands was displaced by the appropriate unlabelled competitor. The pKi for the ligand-receptor combinations are given below.
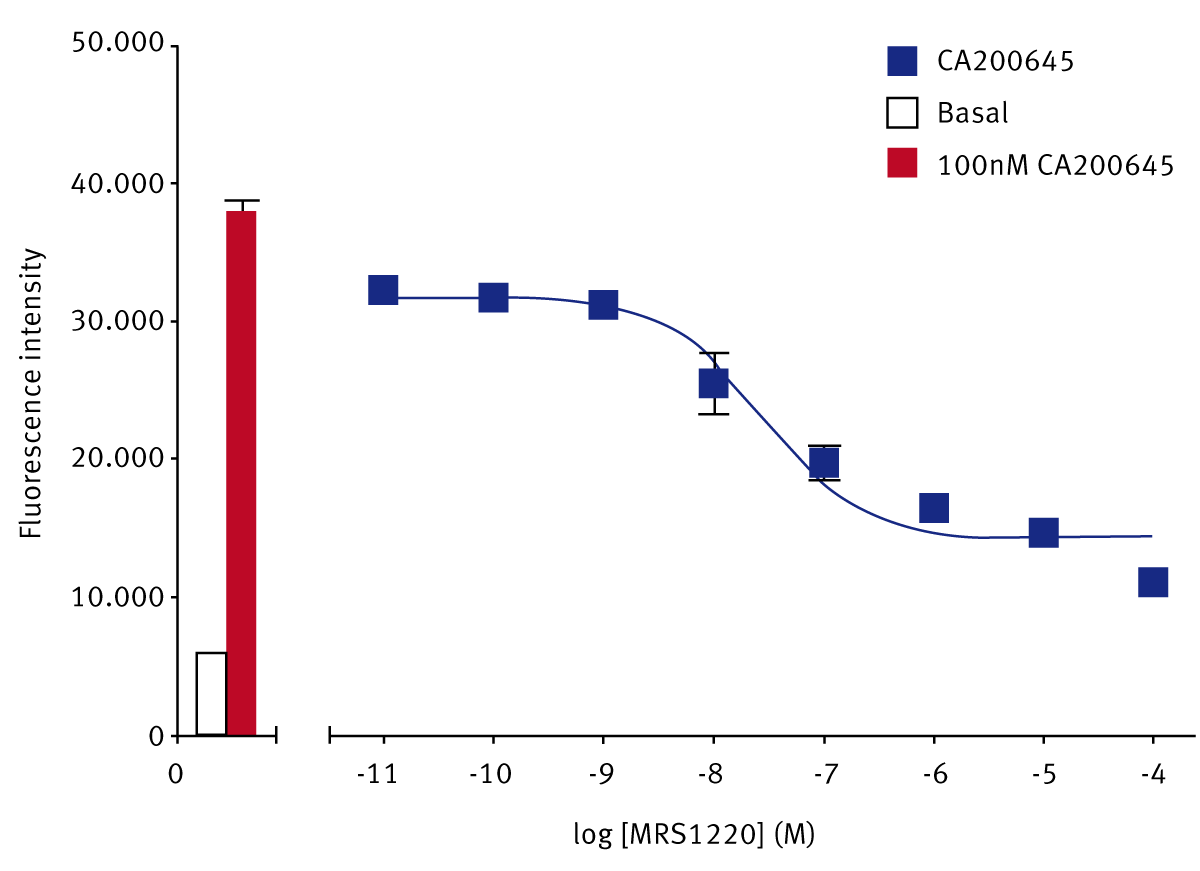
The XAC-derivative adenosine A3 receptor antagonist, CA200645, binding to human adenosine A3 receptors gave a Z’-Factor of 0.62 ± 0.07 (mean ± SEM, n=6). The adenosine A3-selective antagonist MRS1220 displaced the fluorescent ligand, CA200645, with a pKi of 8.58 ± 0.12 (n=5) (Fig. 1). This value is similar to the reported pKi for MRS1220 at the adenosine A3 receptor of 8.76, determined in a radioligand binding assay.
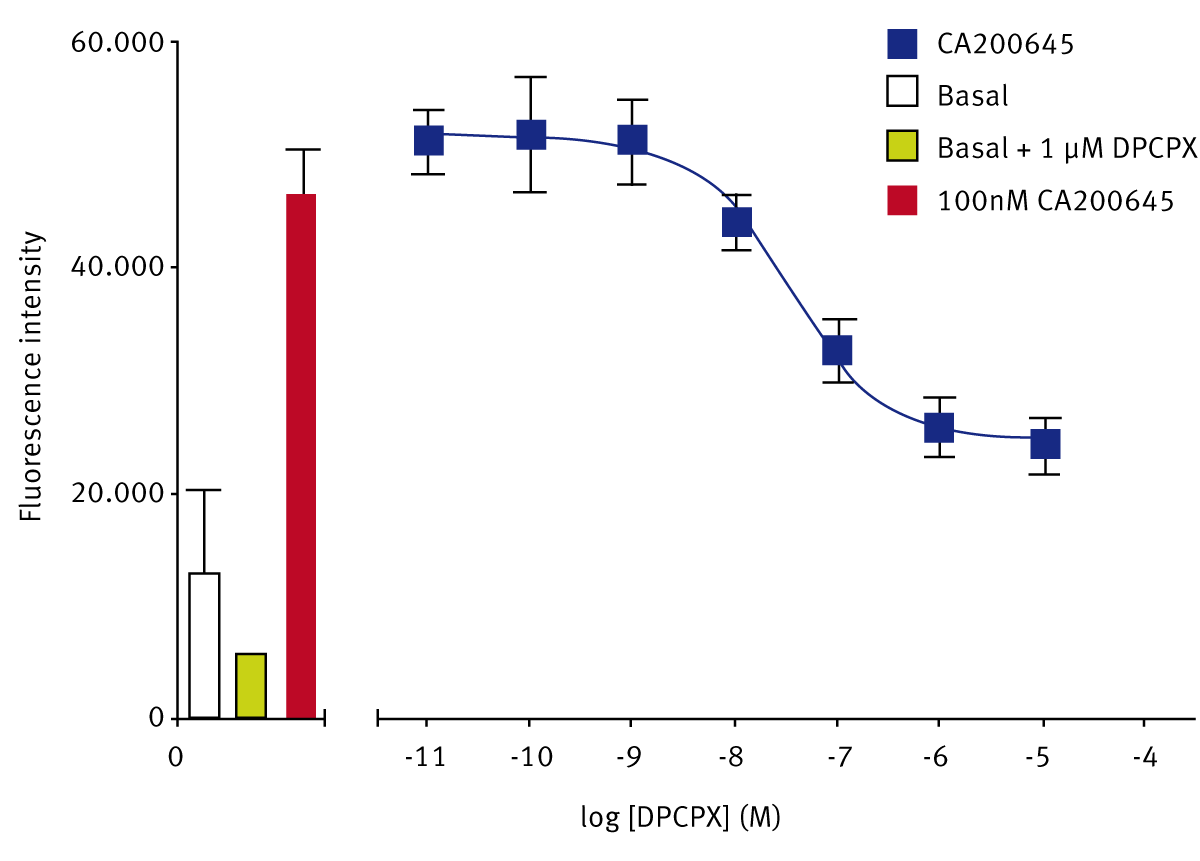
The A3 receptor antagonist, CA200645, bound with lower affinity to adenosine A1 receptors, giving a Z’-Factor of 0.61 ± 0.1 (n=5). The A1-selective antagonist DPCPX was able to displace the A3-selective fluorescent ligand, to give a pKi of 7.47 ± 0.10 (n=4) (Fig. 2). The reported pKi for DPCPX at adenosine A1 receptors is 8.41.
The SKF-83566-derivative dopamine D1 receptor antagonist, CA200767, gave a Z’-Factor of 0.40 ± 0.13 (n=3). The D1-selective antagonist SCH23390 displaced the fluorescent D1 ligand to give a pKi of 8.63 ± 0.03 (n=3) (Fig. 3). The reported pKi for this ligand at the dopamine D1 receptor is between 9.3 and 9.7.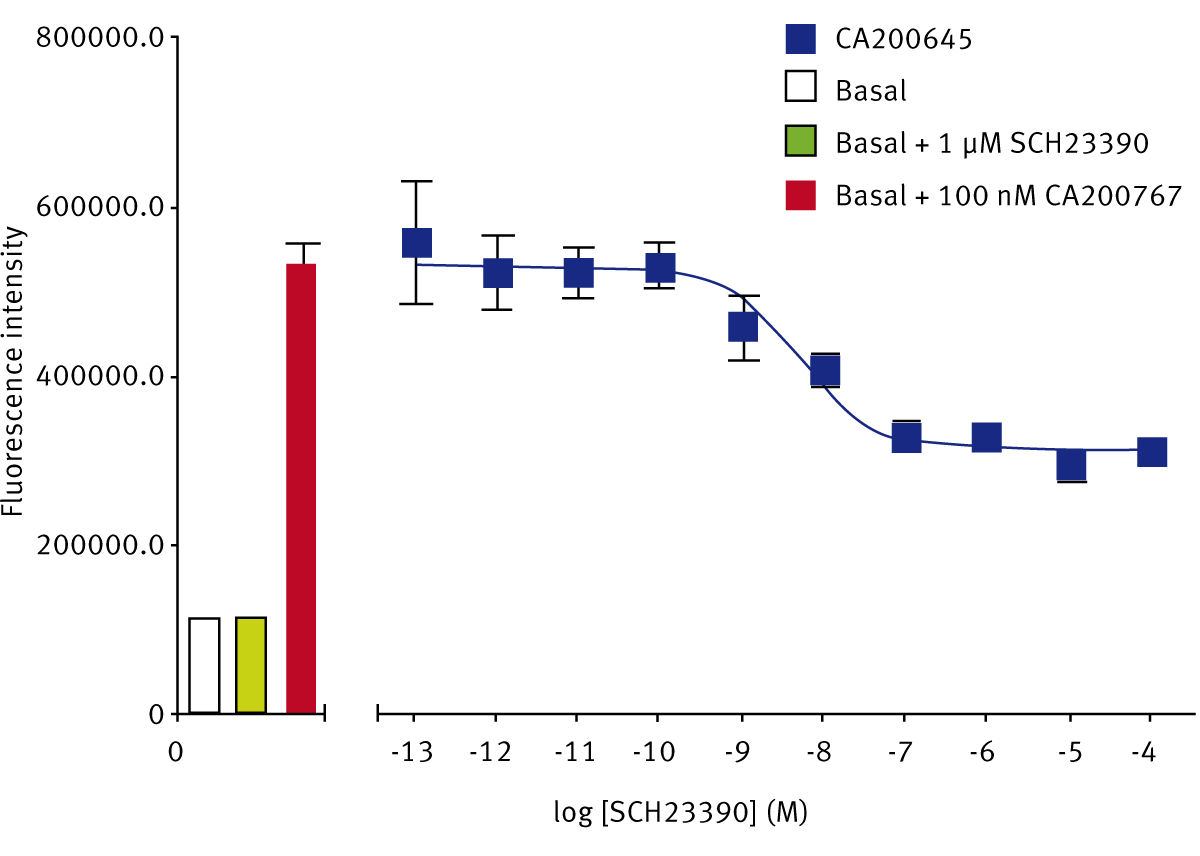
Conclusion
These data illustrate the capability of the PHERAstar FS to rapidly and robustly quantify fluorescent ligand binding to live cells expressing recombinant human GPCRs in 96-well format using a simple ‘add > mix > wash > read’ assay protocol, analogous to whole-cell radioligand binding assays, but with inherent safety and cost advantages. It also offers considerable read and analysis time saving advantages over HCA assays, potentially making this format suitable for high throughput screening (HTS). In addition to the data shown CellAura have also used the PHERAstar FS to multiplex their BODIPY 630/650 ligand binding assay with other fluorophores such as Hoechst or DAPI to normalise for cell number. It may also prove possible to measure down-stream second messenger activation, such as Ca++ using the same cells, to add even more value to the assay data that can be achieved.